1. Introduction
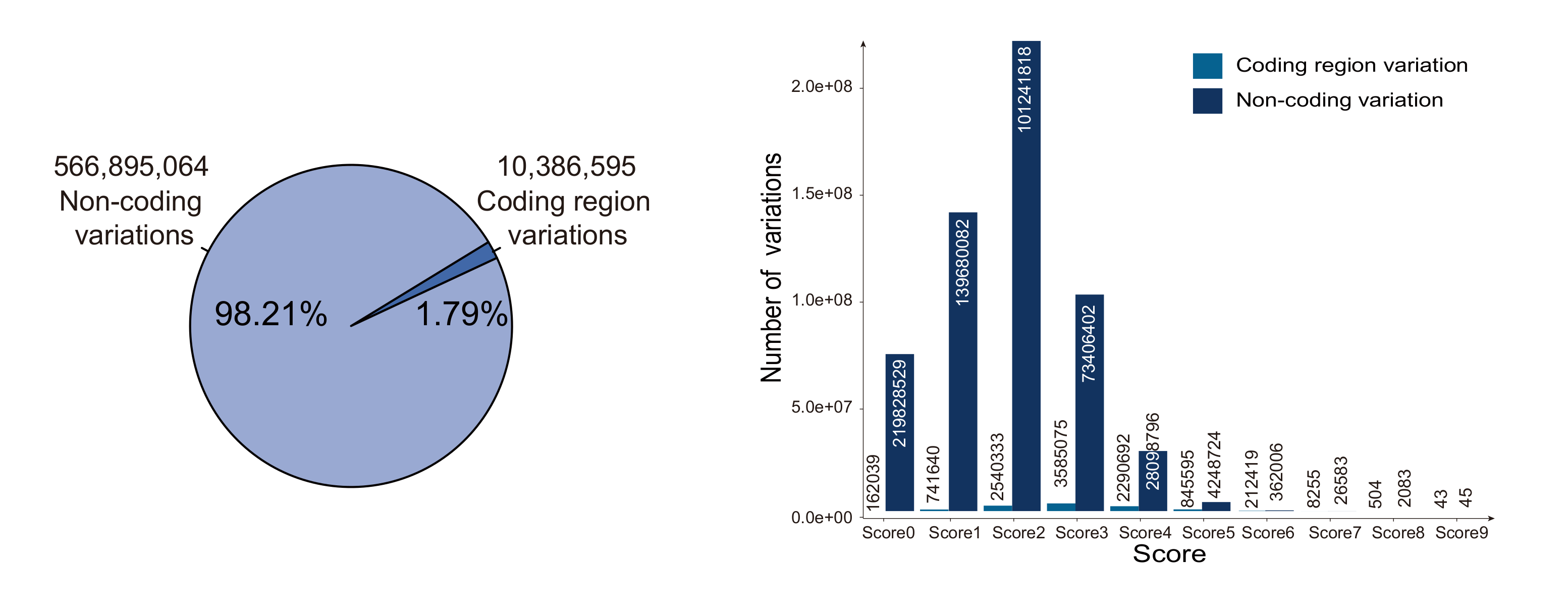
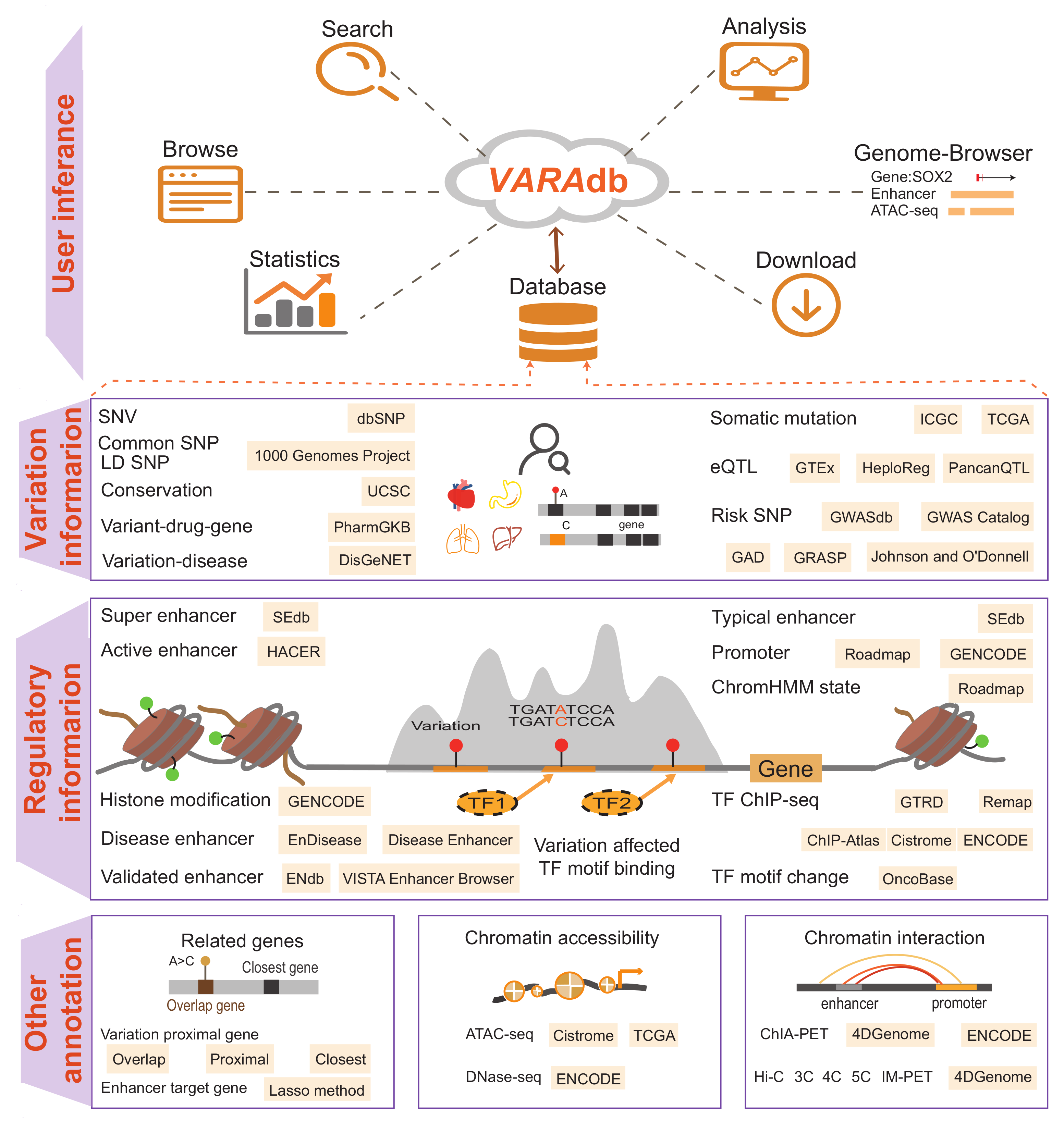
VARAdb is a variation annotation database for human containing 577,283,813 variations. Coding region variations account for ~1.79% of 577,283,813 variations. Among all variations, 10,386,595 are coding region variations and 566,895,064 are non-coding variations and 2,154 are other variations. Among 577,283,813 variations, 10,386,595 are coding region variations, 566,895,064 are non-coding variations. VARAdb provides annotations for the cataloged and novel variations, scores variations based on annotations and performs pathway downstream analysis. The annotations consist of 5 sections including 'Variation information', 'Regulatory information', 'Related genes', 'Chromatin accessibility' and 'Chromatin interaction'. In details, the 'Variation information' section contained risk SNPs, LD SNPs, eQTLs, clinical variant-drug pairs, sequence conservation, motif changes and somatic mutations. In 'Regulatory information' section, VARAdb displays different types of enhancers, promoters, TFs identified by ChIP-seq, ChromHMM core 15 states and histone modifications. The 'Related genes' exhibits variation and variation 3 strategies genes (overlap genes; proximal genes; the closest gene) and enhancer target genes predicted by Lasso method. Accessible chromatin information including ATAC-seq and Dnase-seq is shown in 'Chromatin accessibility' section. In addition, 'Chromatin interaction' section displays Hi-C, ChIA-PET, IM-PET, 3C, 4C and 5C experiment data. VARAdb helps users to select the potential functional variations and develop mechanistic hypotheses of the impact of variations on clinical phenotypes and complex diseases.
2. Content and construction
VARAdb collected a total of 577,283,813 variations not only from dbSNP but also multiple other resources. Notably, 577,098,938 SNVs were collected from dbSNP v151 and 79,482,384 common SNPs were collected from the 1000 Genomes Project. Each common SNP from the 1000 Genomes project has at least one 1000 Genomes population with a minor allele of frequency >= 1%. Millions of LD SNPs of five super-populations (4,477,132 from African; 4,548,152 from Ad Mixed American; 3,693,208 from East Asian; 4,011,947 from European; and 3,838,175 from South Asian) were calculated using phased genotype information accompanying the 1000 Genomes Project phase 3. In addition, we integrated 1,515,001 risk SNPs from the GWAS Catalog, GWASdbv2.0, GAD, Johnson and O'Donnell, and GRASP v2.0. We also obtained 3,998,301 eQTLs from GTEx v7, PancanQTL, and HaploReg v4.1. And VARAdb provided five annotation sections including 'Variation information', 'Regulatory information', 'Related genes', 'Chromatin accessibility' and 'Chromatin interaction'.
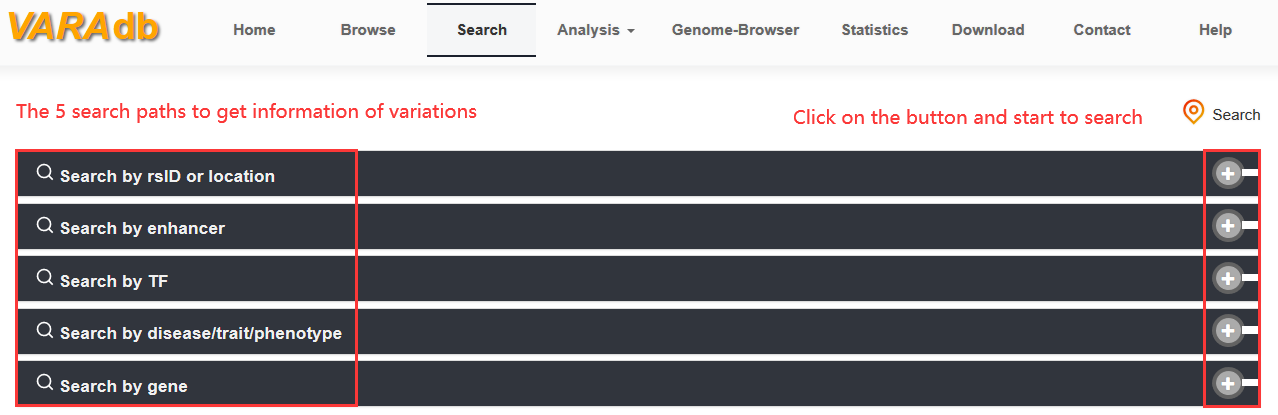
Users can search variations through 5 paths: search by rsID or location, search by enhancer,search by TF, search by disease or trait or phenotype, search by gene as well. In addition, the 'Browse' page has two modules including 'Browse all variations' and 'Browse risk SNPs'. Each module contains an interactive and alphanumerically sort table. When browsing all variations, users can use 'Score', 'Common SNP', 'LD SNP' and 'Variation type' to filter variations of one chromosome. When browsing risk SNPs, users can use 'Source', 'Score', 'Disease/trait/phenotype' to search and filter risk SNPs of interest. Moreover, VARAdb includes analytical tools and genome browser to discover potential biological effects of variations.
3. How to use the VARAdb?
3.1. Search
Users can search variations through five paths including 'Search by rsID or location', 'Search by enhancer', 'Search by TF', 'Search by disease/trait/phenotype' and 'Search by gene'.
3.1.1. Search by rsID or location
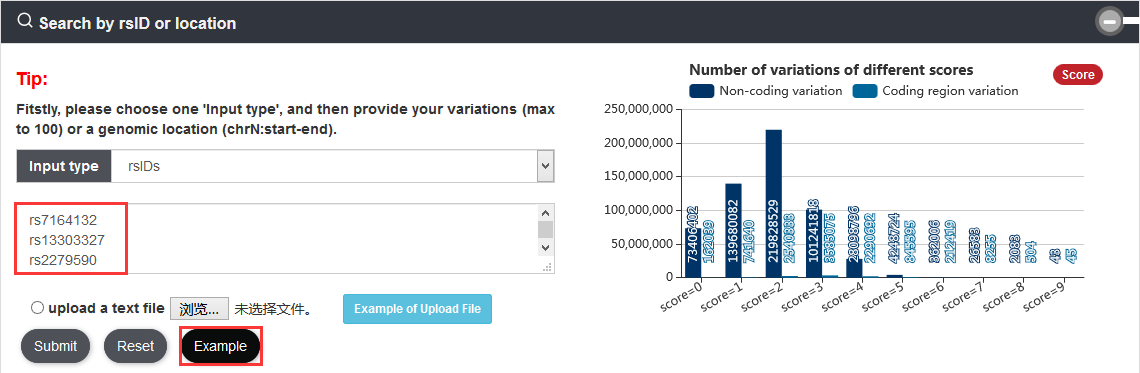
Users can query variations by inputting rsIDs or one genomic location of interest. The search results will be displayed on the next page.
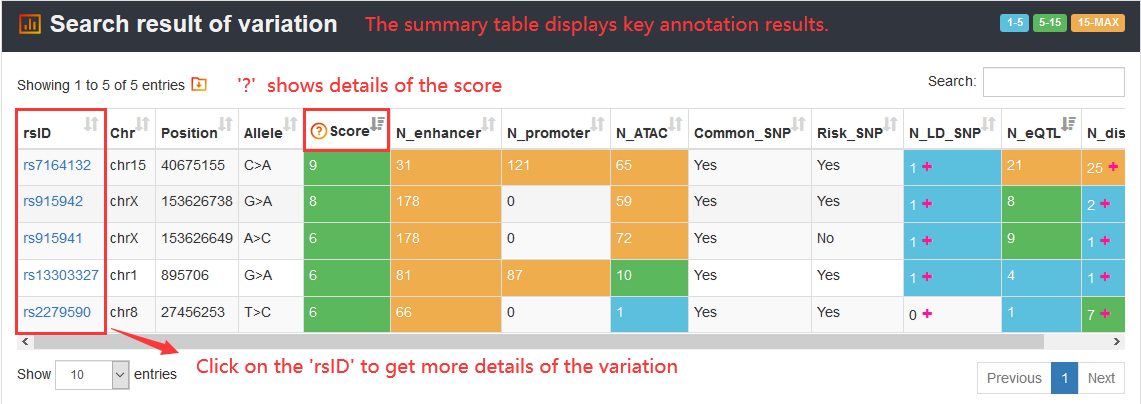

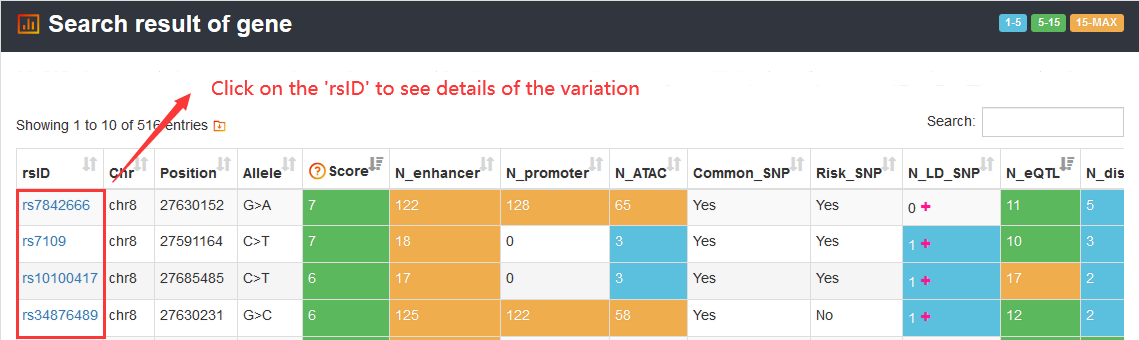
When users click on one 'rsID' of the summary table to view details of a variation, they will get comprehensive information of 5 sections including 'Regulatory information', 'Variation information', 'Related genes', 'Chromatin accessibility' and 'Chromatin interaction'.
Here, we display information of rs7164132, which is scored with 9 in VARAdb. The 'Variation overview' exhibits the summary information of the variation.
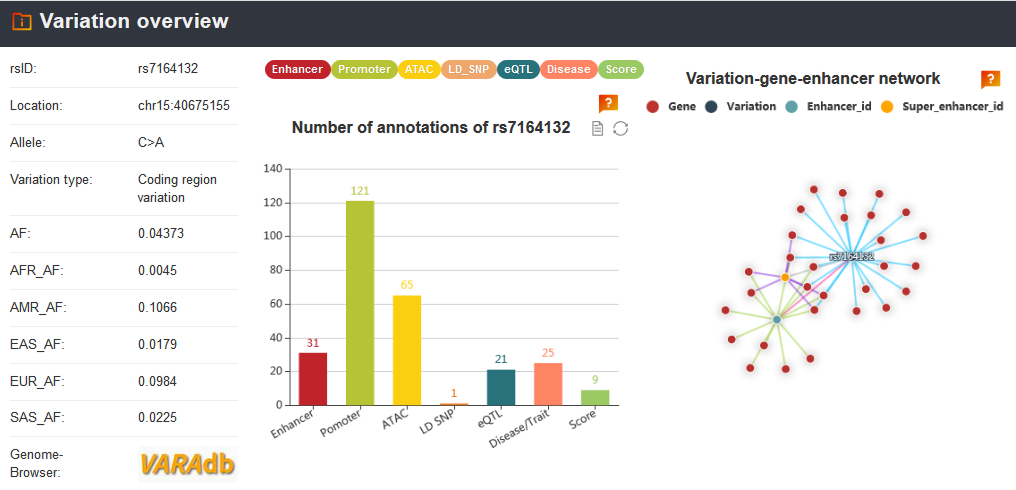
To see regulatory information of the variation, users can focus on 'Regulatory information' section including enhancers, promoters, ChromHMM states, TFs ChIP-seq and histone modifications.
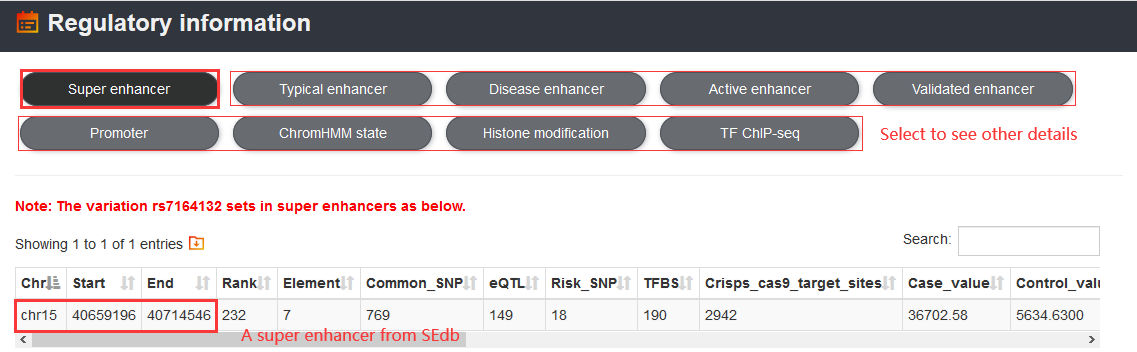
Users can use the 'Variation information' section to access risk SNPs, LD SNPs, eQTLs, TF motif changes, clinical variant-drug-gene pairs, somatic mutations and conservation of the variation.

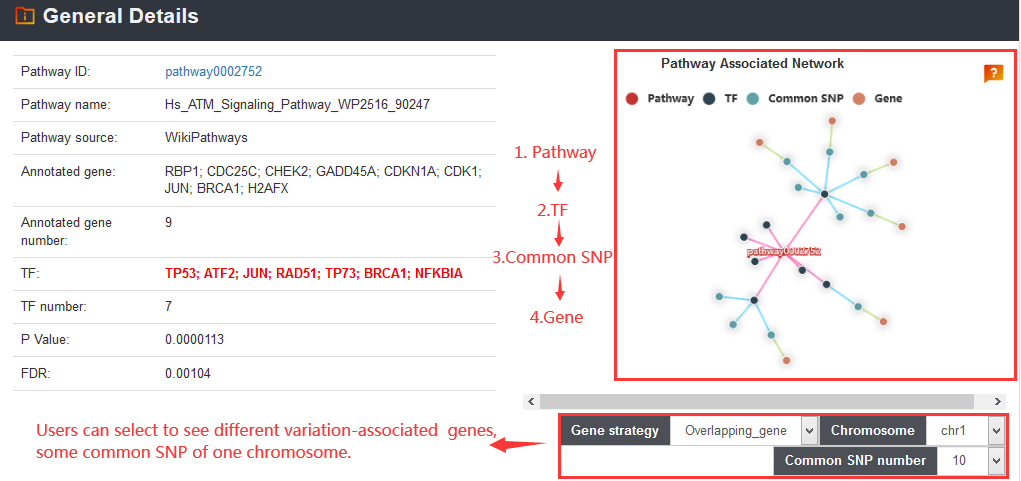
The 'Related genes' section provides variation and enhancer proximal genes and enhancer target genes predicted by Lasso method. VARAdb also provide visualization of some genes according to their genomic location.
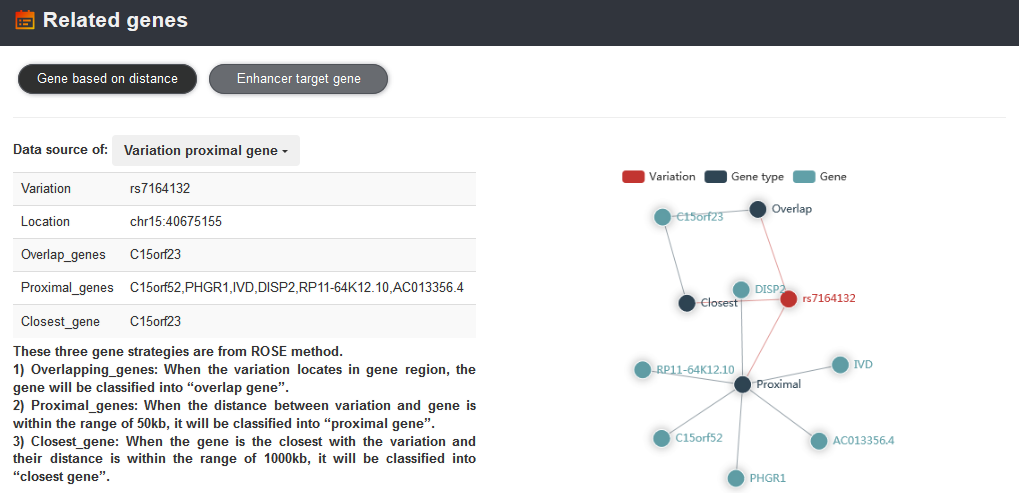

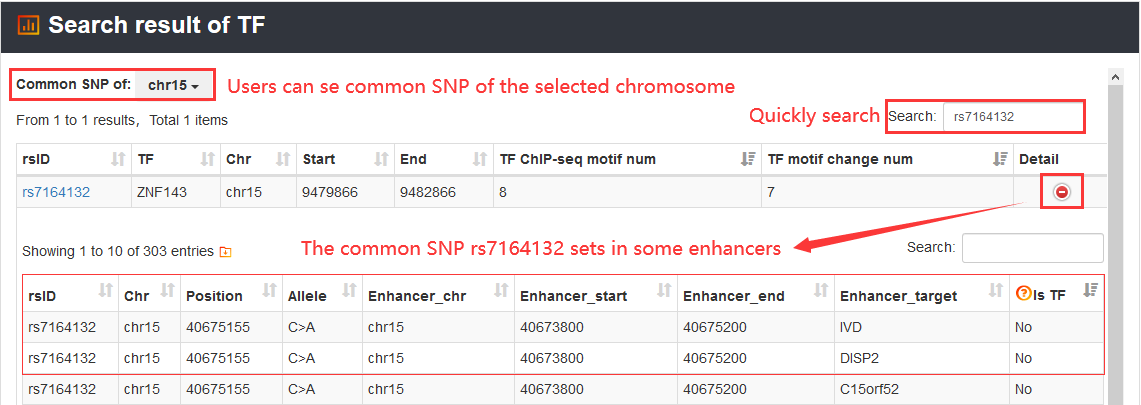
The enhancer regulation with the variation rs7164132 can be viewed.
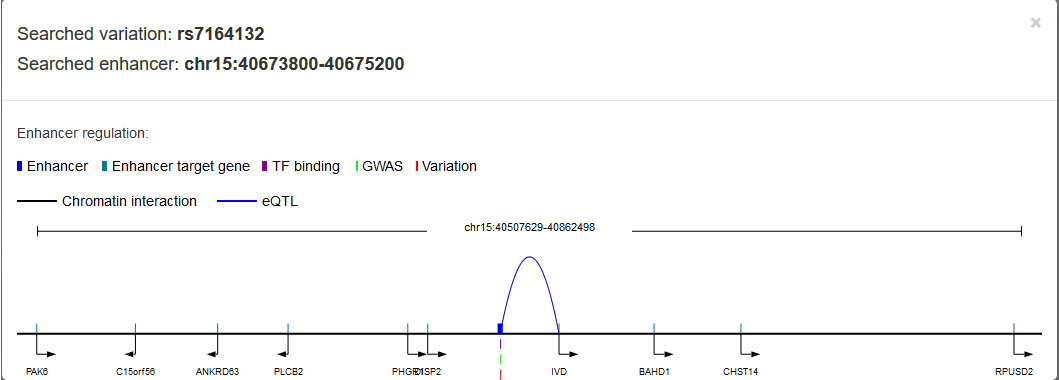
To get accessibility information of the variation, users can use 'Chromatin accessibility' section.
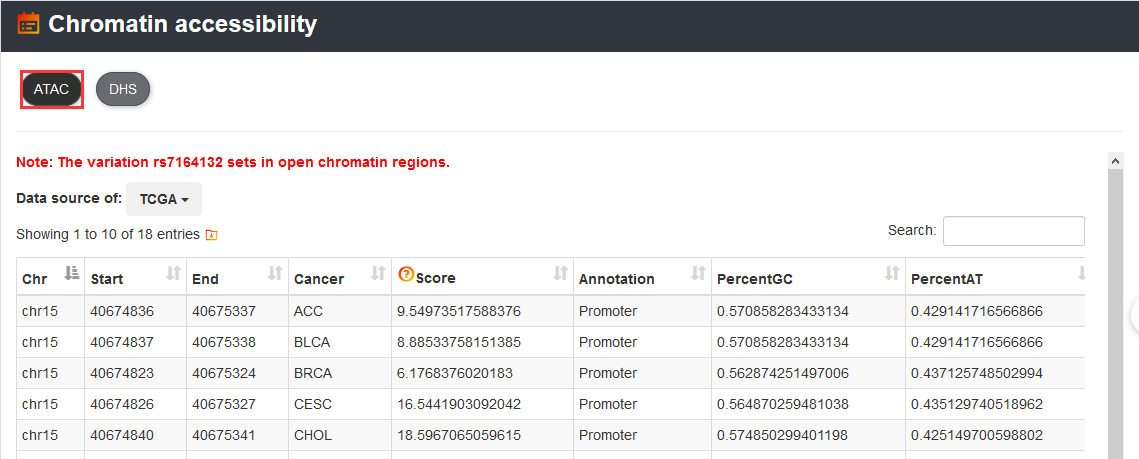
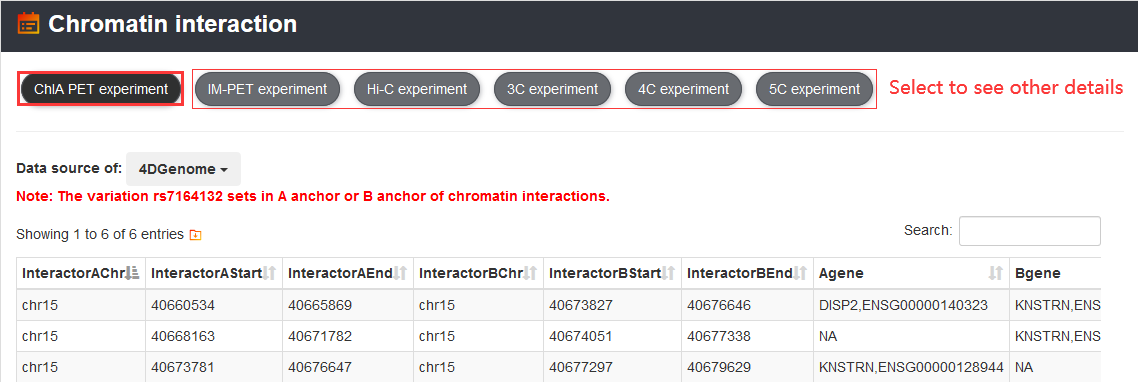
In addition, users can get chromatin interaction information of the variation by 'Chromatin interaction' section, which includes Hi-C, ChIA-PET, IM-PET, 3C, 4C and 5C experiment data.
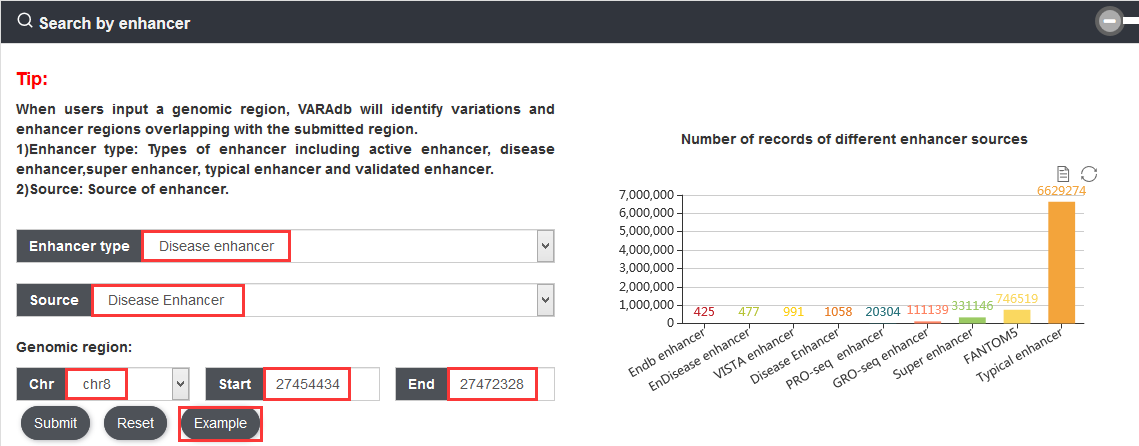
3.1.2. Search by enhancer
Users can query by inputting a genomic location of an enhancer and selecting one of the enhancer data sources. The search result will be displayed on the next page.

When users click on 'Enhancer_id', VARAdb will display variations that are set in the selected enhancer.

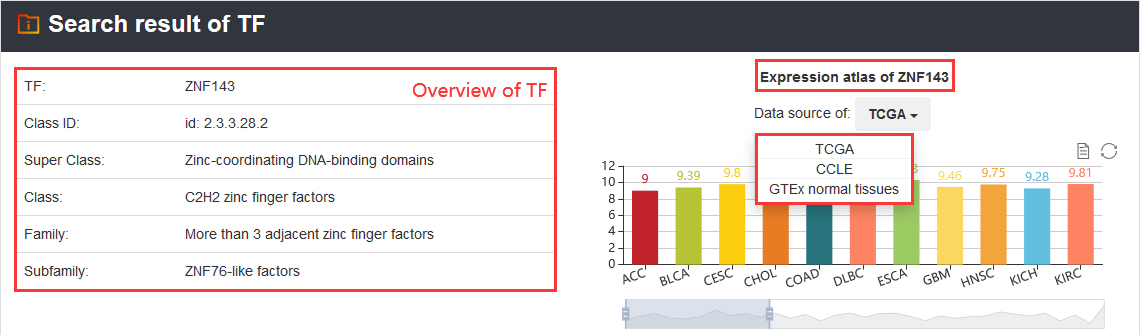
3.1.3. Search by TF
Users can query by inputting a TF. The search results will be displayed on the next page.

3.1.4. Search by disease
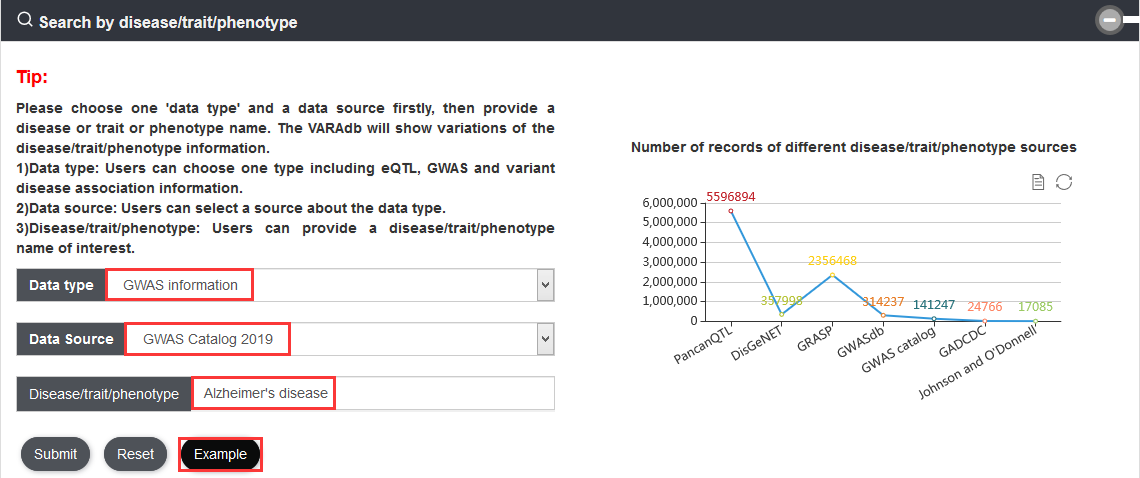
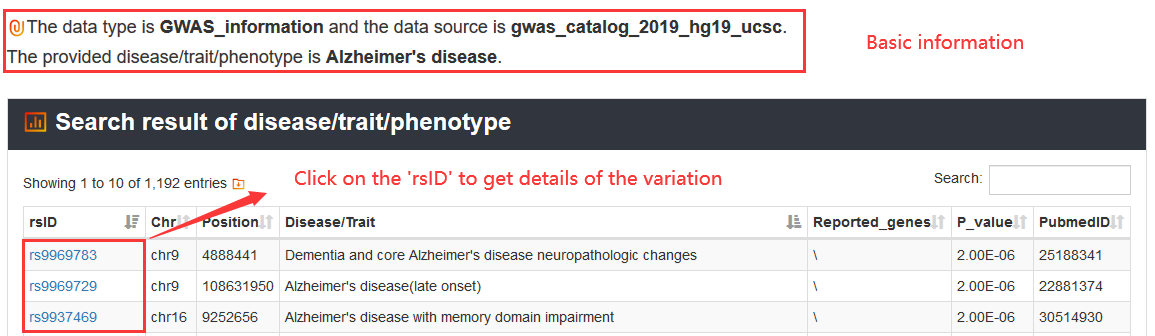
Users can query by inputting a disease/trait/phenotype name and select one of the data sources. The search result will be displayed on the next page.
3.1.5. Search by gene
When users input a gene, VARAdb will identify variations associated with the gene. Note that the gene is one of the eQTL genes,gwas reported genes or the closest gene of variation.
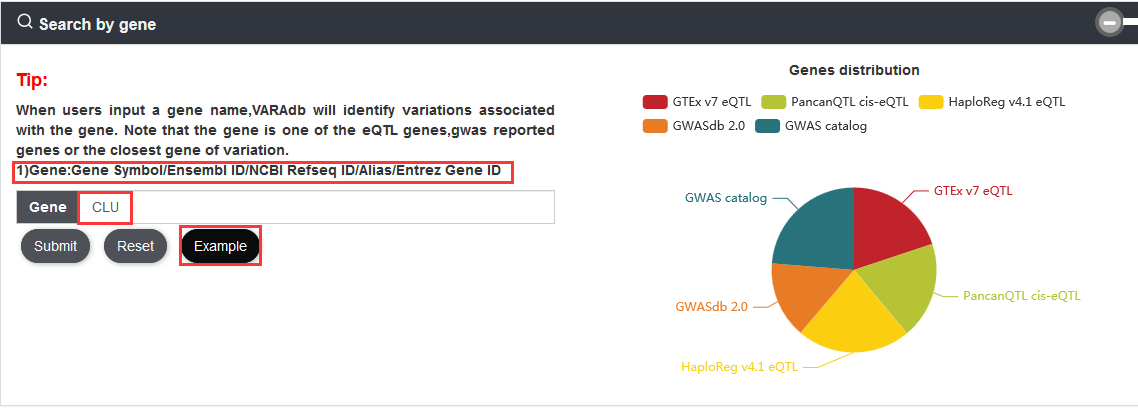
3.2. Browse
In order to make users browse all variations more conveniently and quickly, we have designed two modules including 'Browse all variations' and 'Browse risk SNPs'. The 'Browse' page owns an interactive and alpha numerically sort table, from which users can use different conditions to filter variations and risk SNPs. When browsing all variations, users select at least one chromosome to browse variations and use 'Score', 'Common SNP', 'Risk SNP' and 'Variation type'. In addition, risk SNPs showed close relationships with different diseases or traits or phenotypes. When browsing risk SNPs, users can filter risk SNPs using 'Source', 'Score', 'Disease/trait/phenotype' to search and filter risk SNPs with different scores in VARAdb. For each risk SNP, its score ranges from 6 to 9 and was calculated based on its annotation categories as previously described above. The 'Show entries' drop-down menu is provided to change the number of records per page. In addition, users can view the details of risk SNP by clicking on 'rsID'.

3.3. Analysis
3.3.1. Novel variant annotation
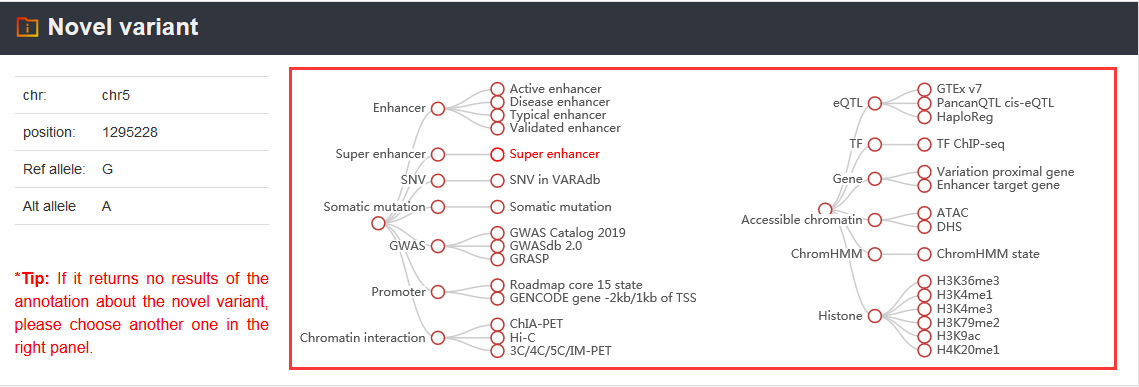
For the novel variant of interest, we designed a ‘Novel variant annotation’ analysis to provide 5 annotation sections. In details, information includes accessible chromatin, ChromHMM state, histone modification, transcription factor, chromatin interaction, super-enhancer, promoter, gene, eQTL, GWAS, somatic mutation and SNV. Users can investigate the potential functions of the novel variant according to annotation results in VARAdb with friendly interface. Users can not only manually input many novel variants (one variant with chrN position ref_allele alt_allele per line) but also upload a set of variants in a text file or a VCF file.
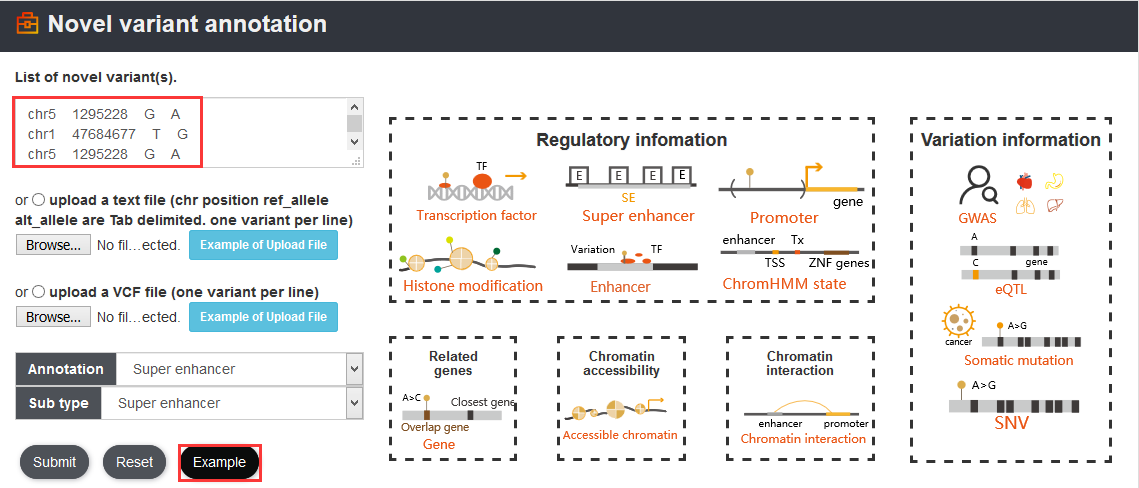
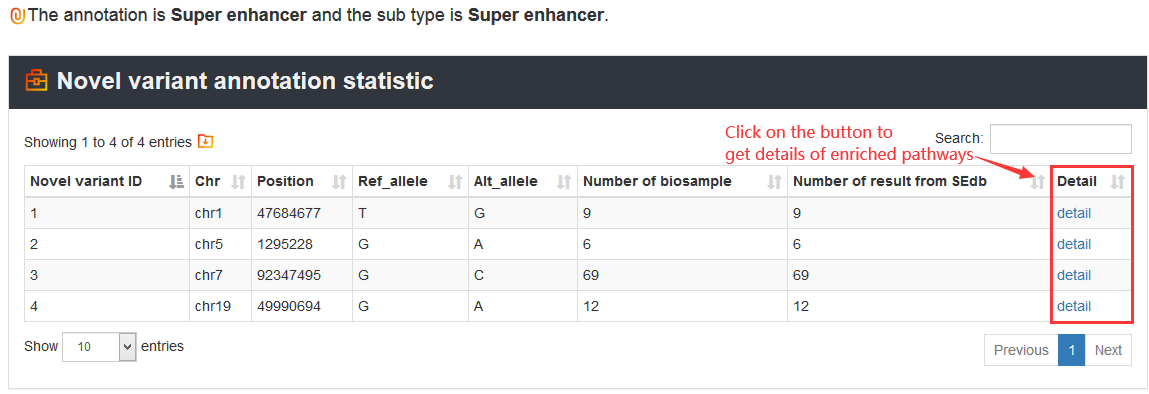
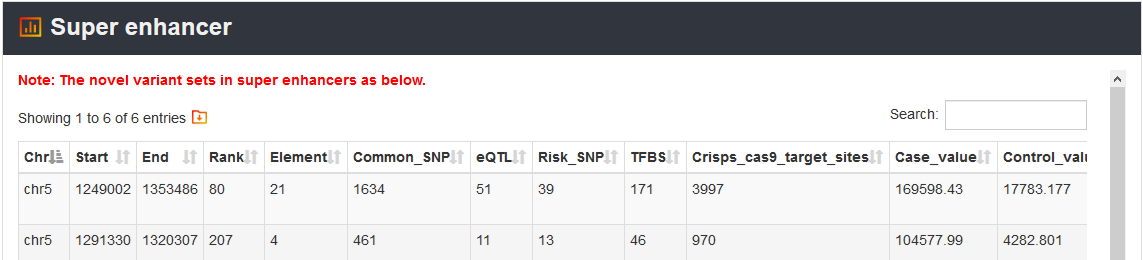
Here, we provided a TERT promoter mutation (chr5:1295228:G>A) as an example. Users can click Analysis→Novel variant annotation→chr5→1295228→G→A→Super enhancer, the variant’s analysis results will be displayed on the next page.
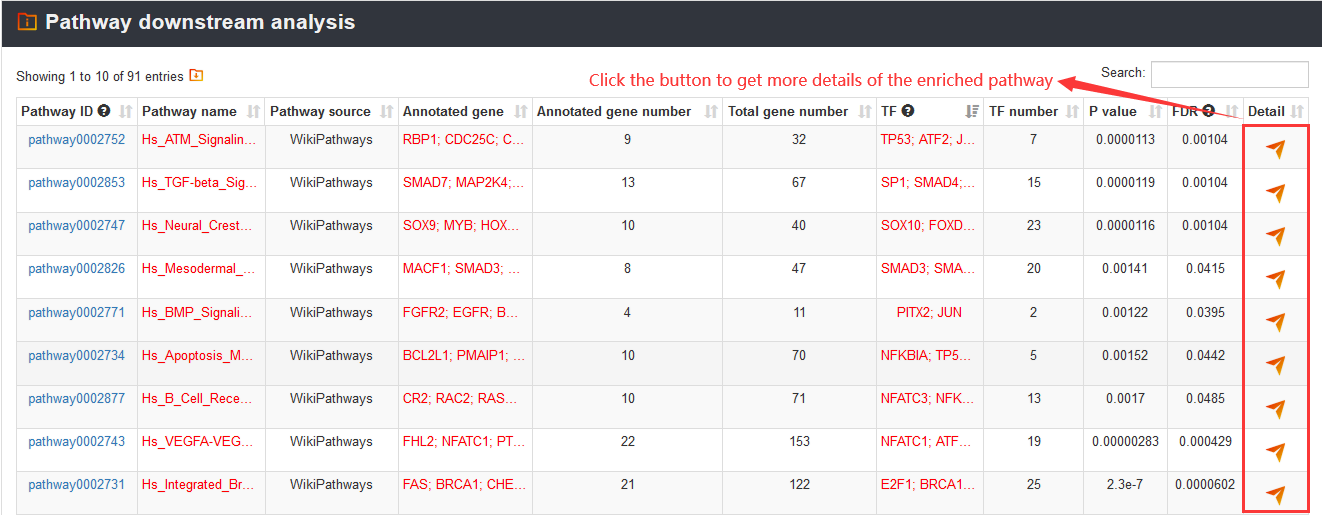
3.3.2. Pathway downstream analysis
To analyze complex regulatory networks formed by pathways, transcription factors (TFs), common SNPs, and common SNP-associated genes, users can input a gene set of interest and select a pathway database to perform pathway downstream analysis. Users can not only manually input genes (one gene symbol per line) but also upload a gene set in a text file. In addition, VARAdb provided an apparent introduction of the function at the bottom of the database. When analyzing, VARAdb will provide different indications explaining why there is no result.
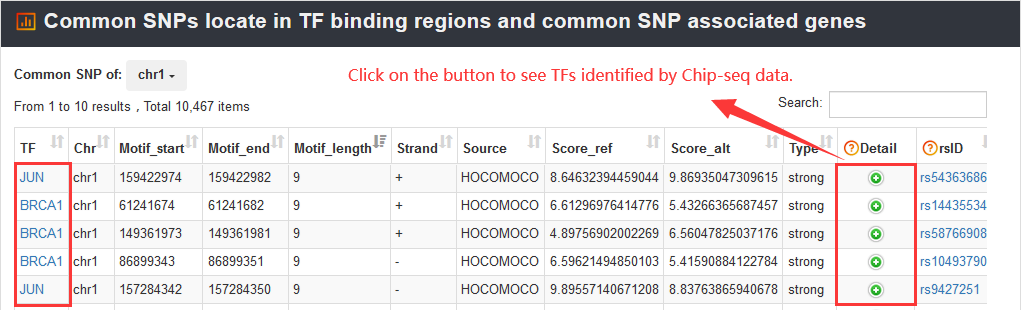
And VARAdb will identify significantly enriched pathways (calculated using hypergeometric test), downstream TFs, common SNPs affected TF motifs binding and common SNP-associated genes. In addition, the false discovery rate (FDR) is also calculated to correct for multiple testing.
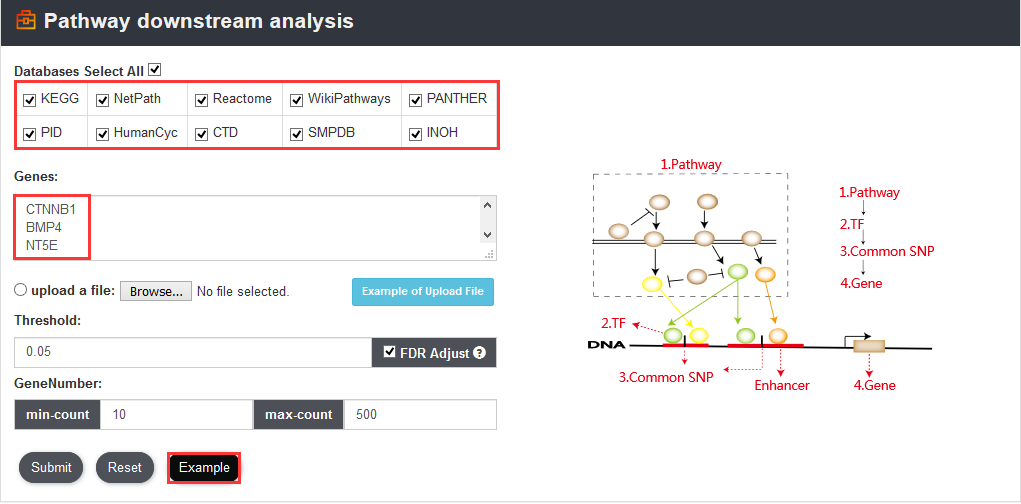
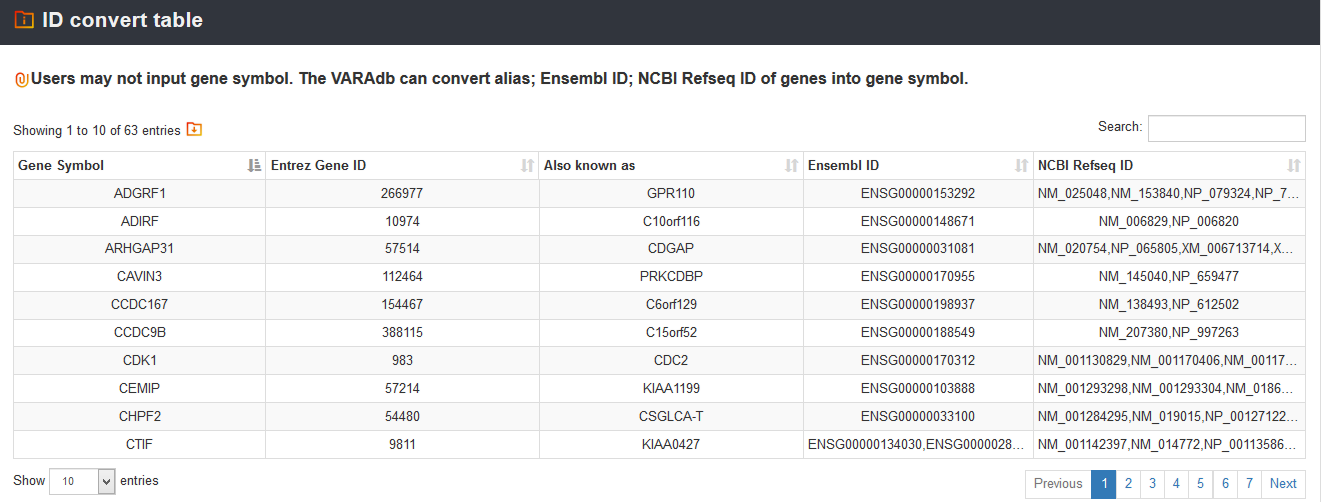
If users input other gene ID or alias, VARAdb can help to convert to gene symbol name.
The VARAdb will show the output table containing summary information of enriched pathways including Pathway ID, Pathway name, Pathway source, Annotated gene, Annotated gene number, Total gene number, TF and TF number, P value and FDR. Furthermore, detailed description of the regulatory networks can be obtained by clicking the 'Detail' button.
3.4 Genome-Browser
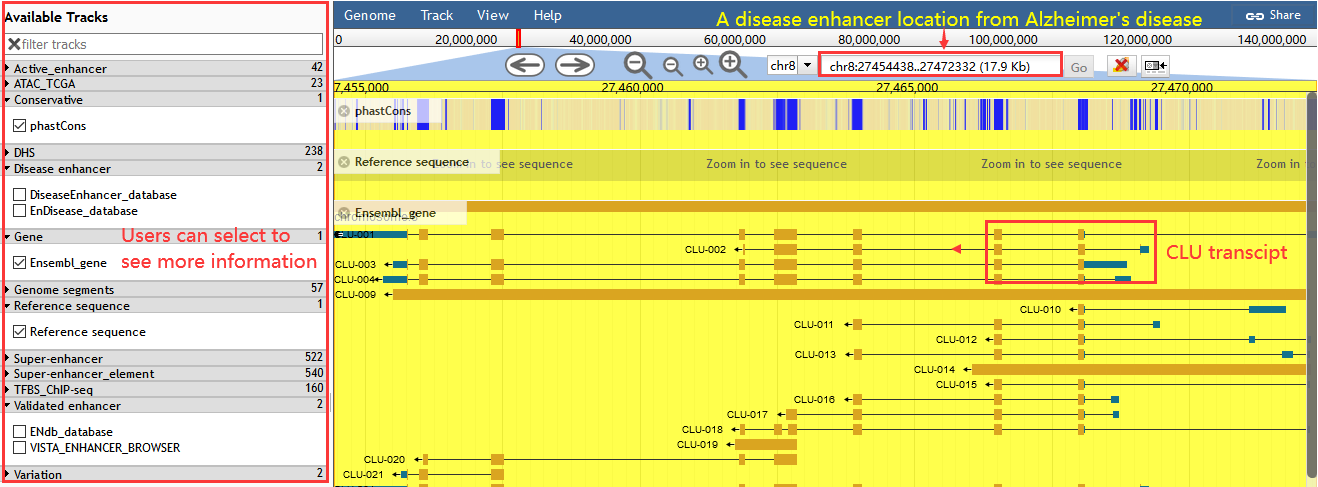
Users can view regulatory information of variations in the genome by using 'Genome-Browser' page. Through useful tracks, users can get information including common SNPs, conservation score, enhancers, super enhancers, nearby genes, TFBS by ChIP-seq, genome segments, ATAC accessible chromatin regions and DHSs.
3.5 Download
The ‘Download’ page exhibits ‘Variation information’, ‘Regulatory information’, ‘Variation related genes’, ‘Chromatin accessibility’ and ‘Chromatin interaction’ data for users to download. Moreover, the detailed description of file is also displayed.
4. Development environment
Using MySQL 5.7.27, we developed the current version of VARAdb that runs on a Linux-based Apache web server. We utilized PHP 5.6.40 for sever-side scripting, Bootstrap v3.37 and JQuerry v2.1.1 for interactive interface building, Echats for visualization and JBrowser for genome browser. To display best, we recommend using a comprehensive web sever that supports HTML5 standard, for example, Firefox, Google Chrome and Safari.
The research community can access information freely in VARAdb database without registering or logging in. The web link of VARAdb is http://www.licpathway.net/VARAdb.
5. Process of updating the database
In the process of developing the database, we have fully considered the process of updating the database. Because there are many data sources, we designed multiple sections and data modules and managed updates in a modular way. This will effectively ensure the security and controllability of data and improve the efficiency of updating the database. Specifically, we designed five annotation sections including ‘Variation information’, ‘Regulatory information’, ‘Related genes’, ‘Chromatin accessibility’ and ‘Chromatin interaction’. For each section, we divided many sources of the same or similar characters into a group, which is as an independent module. Due to storing tables based on the individual module, each module can work and be updated relatively independently. We developed a process of updating the database. When updating each data source, the quality of data will be controlled according to the modular process. If meeting a series of requirements, the data will be updated.
